Principles and Applications of Modern
DNA Sequencing
EEEB GU4055
Session 14: Phylogenomics
Today's topics
1. Phylogenomics introduction
2. The coalescent and why we do phylogenomics
3. Coalescent simulation (exercise)
4. Subsampling methods: anchored hybrid enrichment
5. Subsampling methods: RAD-seq (exercise)
Phylogenomic sampling
Characterize evolutionary history from a subset of sampled genomes (individuals).


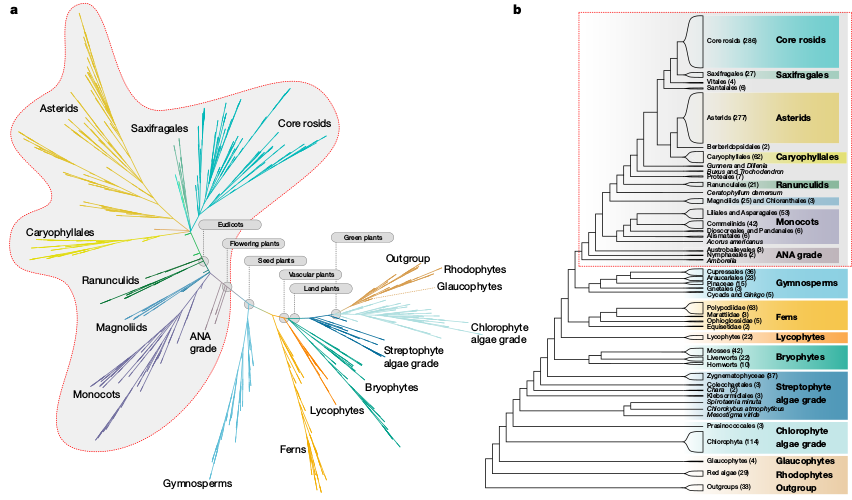

Phylogenomic sampling
Characterize whole genomes from a subset of sequenced markers.

Genealogical variation
It is important to examine evolutionary history across the entire genome.

Historical introgression/admixture
It is important to examine evolutionary history across the entire genome.

The Coalescent
A model that describes the expected waiting time until two or more
samples share a most recent common ancestor. The distribution of
coalescent times within a population, or between populations, provides information about their history.
There are many genealogical histories that could possibly explain the genetic relatedness of a set of samples. We cannot observe the genalogies directly, only the sequence data that evolved on those genealogies.
Coalescent simulations provide a means to ask: "can the genetic variation that I observe in my samples be explained by neutral evolutionary processes?"
Population parameters (Ne)
The effective population size (Ne) of a population describes the probability that two samples share a common ancestor in the previous generation. This parameter does not translate directly to the actual population size, though they are likely correlated. Other factors like non-random mating and population structure also affect Ne.
Single population model
If we assume that a population is randomly mating (panmictic) and neutrally evolving then the expected waiting time until n samples coalesce can be modeled entirely by Ne.
Because n samples can share many possible genealogical histories (remember how big tree space is), and their genealogical relationships are expected to vary across their genomes (recombination makes different regions independent of others), we expect to observe a large variation in genealogical histories when examining many loci for n samples.
The coalescent model treats genealogies as a random varaible. We are interested in the expected distribution of variation when integrating over many genealogies.
Multiple population (structured) coalescent
When modeling multiple populations a "species tree" topology (e.g., "Species Tree") defines when different samples or their ancestors are able to share a parent in a previous generation.
To predict the expected genetic similarity of samples in a structured coalescent model requires estimating Ne for each lineage as well as T, the divergence time of the populations.
Modern phylogenetic inference methods are based on the multispecies coalescent model which calculates the likelihood of observed genetic data given a set of parameters: Ne, T, and a topology. Searching over many topologies and many parameters can identify a best species tree model that explains variation among genealogies.


Rokas et al.: Discussion
- What type of sequence data did they use?
- Is this a shallow or deep phylogenetic question?
- Why is there so much variation among gene trees?
- What is their recommended solution (in 2002)?
- Is this still a recommended method?
- Would sampling more data help infer a better tree?

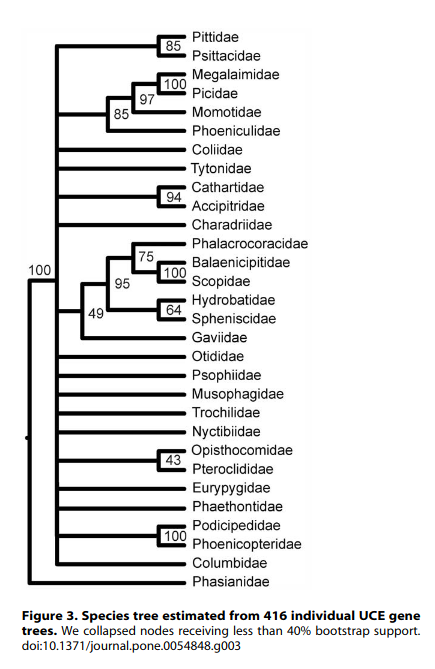
McCormack et al.: Discussion
- What type of sequence data did they use?
- Is this a shallow or deep phylogenetic question?
- Is there agreement among the gene trees?
- Is their species tree highly supported?
- Would sampling more data help infer a better tree?
Phylogenomic inference methods

Challenges: missing data

Preparing Genomic Libraries
Wet lab techniques for taking extracted DNA and ligating synethesized nucleotides to it to prepare it for a sequencing machine.
Adapter sequences are oligonucleotides with a sequence that binds to some feature of the sequencing machine.
Barcodes/Indices are unique molecular identifiers that can be ligated (attached) to DNA fragments so that they can be pooled for sequencing and later assigned to different samples based on the barcode (demultiplexed).
Targeted Hybrid Enrichment Methods
Methods for subsampling the genome to select particular regions for sequencing. Requires a priori knowledge about sequence at the regions of interest.
Design and order synethesized RNA baits that will bind to target DNA region. These baits are ligated to magnetic beads that allow them to be pulled out of solution with powerful magnets. This will enrich the DNA sample for the targeted regions.
Shotgun sequence the enriched library and assemble reads into contigs overlapping the targeted region.
Exome sequencing (WES)
The exome is composed of all of the exons within the genome. It is different from the transcriptome, which contains all RNA transcribed in a cell. The transcriptome will vary among different cell types whereas the exome does not.
Targeted exome sequencing uses hybrid target capture to enrich a DNA extraction for coding regions before shotgun sequencing. It requires a priori knowledge of the gene sequences.
Whole Exome Sequencing is mostly used in human biomedical research, and model organism research, since designing an array or probe set for one species requires a high quality reference genome and is costly (i.e., needs to be used many times to recoup costs).
Anchored hybrid enrichment methods
For phylogenomic analyses we typically do not need the whole exome, and instead design baits for just a subset of exons. In particular, exons that are highly conserved and occur as a single copy (not duplicated). RNA baits can be designed for many closely related taxa based on one or more closely related genomes. If the samples differ too much from the taxon used for bait design you end up with missing data.
Ultraconserved Elements
Some genomic regions have been identified that are very very highly conserved among even very divergent taxa (e.g., all birds or all mammals). Sometimes these regions have unknown functions, some are related to important developmental genes.
Baits have been designed that target these UCE regions and extend away from them for several hundred base pairs. The center has almost no variation but on the ends of contigs more variation is detected.
Whereas it is often very hard to align orthologous regions among very distantly related species, UCEs seem to work well for obtaining many hundreds or thousands of orthologs.

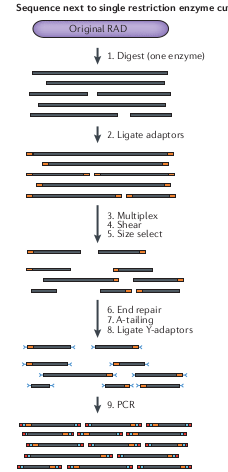
RAD-seq
Subsample many thousands of regions across the genome without need to design baits. Fast and efficient subsampling method.
Initially used for association mapping, and genetic maps, where sparsely spaced markers are sufficient to identify ancestry relative to parents.
But because it is easy to generate thousands of markers it also became popular for population genetic and phylogenetic analyses.
Drawbacks of RAD-seq
- Distantly related samples will not share the same restriction recognition sites (e.g., they accumulate mutations) and so it is characterized by a lot of missing data
- For organisms with small genomes it is increasingly affordable for many types of questions to simply shotgun sequence the whole genome.