Notebook 3.2: Gene transcription and translation¶
This notebook is associated with the following accompanied reading:
- Yandell, Mark, and Daniel Ence. 2012. “A Beginner’s Guide to Eukaryotic Genome Annotation.” Nature Reviews Genetics 13 (5): 329–42. https://doi.org/10.1038/nrg3174.
- [Optional] additional reading on the GFF file format and how it encodes genomic information: The Sequence Ontology.
Learning objectives:¶
By the end of this module you should be able to:
- Use a Python dictionary to represent the genetic code.
- Desribe how genes, mRNA transcipts, and proteins are related.
- Synthetically translate and transcribe genes using Python.
Introduction to gene expression¶
Genomes contain thousands of genes, as well as many other regions that do not contain genes and may or may not have some other function. Genes are often of special interest as they are the units that encode the information for proteins, and therefore for much of the primary biological functions in organisms. The central dogma of molecular biology which relates how information is stored and transferred between DNA, RNA, and proteins was discovered shortly after the discovery that DNA itself was the molecule of genetic information storage.
The DNA sequence of genes provides the underlying information for gene transcription, the first step of gene expression. DNA determines the starting and ending sites, and is a template for the RNA sequence that will be produced. DNA itself simply holds this information in the sequence of its bases. Proteins do most of the actual work of transcribing genes by recognizing certain regions of DNA to bind to, and synthesizing RNA based on the sequence.
Messenger RNA (mRNA) transcripts are a type of RNA that encodes for an amino acid sequence of a polypeptide (a protein, generally). The codon sequence in mRNA is composed of sets of three bases at a time, starting from a special codon that initiates the sequence called a start codon, and ends at a codon that signals the end termed a stop codon. The sequence of codons will be translated into an amino acid sequence that will fold to form the protein.
The genetic code:¶
We can easily represent the genetic code for translation using a Python dictionary. Here the keys are codons (sets of three bases) and the values are amino acid residues (represented by the shorthand single letter codes: e.g., L = leucine). We can query this dictionary object with a codon "CTA" and it will return the amino acid "L".
GENCODE = {
'ATA': 'I', 'ATC': 'I', 'ATT': 'I', 'ATG': 'M',
'ACA': 'T', 'ACC': 'T', 'ACG': 'T', 'ACT': 'T',
'AAC': 'N', 'AAT': 'N', 'AAA': 'K', 'AAG': 'K',
'AGC': 'S', 'AGT': 'S', 'AGA': 'R', 'AGG': 'R',
'CTA': 'L', 'CTC': 'L', 'CTG': 'L', 'CTT': 'L',
'CCA': 'P', 'CCC': 'P', 'CCG': 'P', 'CCT': 'P',
'CAC': 'H', 'CAT': 'H', 'CAA': 'Q', 'CAG': 'Q',
'CGA': 'R', 'CGC': 'R', 'CGG': 'R', 'CGT': 'R',
'GTA': 'V', 'GTC': 'V', 'GTG': 'V', 'GTT': 'V',
'GCA': 'A', 'GCC': 'A', 'GCG': 'A', 'GCT': 'A',
'GAC': 'D', 'GAT': 'D', 'GAA': 'E', 'GAG': 'E',
'GGA': 'G', 'GGC': 'G', 'GGG': 'G', 'GGT': 'G',
'TCA': 'S', 'TCC': 'S', 'TCG': 'S', 'TCT': 'S',
'TTC': 'F', 'TTT': 'F', 'TTA': 'L', 'TTG': 'L',
'TAC': 'Y', 'TAT': 'Y', 'TAA': '_', 'TAG': '_',
'TGC': 'C', 'TGT': 'C', 'TGA': '_', 'TGG': 'W',
}
GENCODE["CTA"]
A biological example¶
Although transcription and translation of genetic information takes place entirely through the interactions of molecules (amazingly), it is still simply a transfer and translation of information; as such, we can emulate it using digital information, and code. Let's transcribe and translate an example portion of a genome sequence that contains a gene using some example Python code. We'll start by generating the sequence itself.
# we'll use the random function
import random
def random_dna(length):
"returns a string of randomly drawn bases of A,C,T,G"
seq = "".join([random.choice("ACGT") for i in range(length)])
return seq
random_dna(20)
def random_coding_dna(length):
"""
returns a string of (length) joined random triplets of bases
(codons) not including start or stop codons.
"""
# empty string to fill
seq = ""
# list of all codons except start and stops
codons = [
i for i in GENCODE.keys() if i not in
["ATG", "TGA", "TAA", "TAG"]
]
# fill string with n random codons
seq = "".join([random.choice(codons) for i in range(length)])
return seq
random_coding_dna(20)
Start and stop codons¶
The primary start codon in eukaryotes is "AUG" (Ts are replaced with "U" in mRNA), which is "ATG" in DNA, and translates to methionine. There are three stop codons, UGA, UAA, and UAG ("TGA", "TAA", and "TAG" in DNA sequence). You can see in the GENCODE dictionary above these do not translate to an amino acid but instead stop translation. We'll add a start and stop codon to our sequence as well. We're now ready to simulate a genomic region.
Our aim is to simulate a region that looks like this:
----non-coding-region----|-----coding-region------|-----non-coding-region-------
(ATG) (TGA)Here we expect that in the coding region, between the ATG and TGA codons, all of the DNA will be in phase so that it is composed of a series of codons that will not terminate by a stop codon too early. By contrast, in the non-coding region it is just random DNA and there may be sequences that by chance look like a stop or start codon, but they are not actually transcribed or translated. We can simulate this by combining string objects for each part of the DNA.
# use a random seed so we will all draw the same random values each time (repeatable)
random.seed(12345)
# create a genomic region containing a coding region surrounded by non-coding DNA
region = random_dna(300) + "ATG" + random_coding_dna(300) + "TGA" + random_dna(300)
region
Explore transcription with Python dictionaries¶
OK, now that we have a genomic region that should contain a gene, let's try to extract the gene region from the DNA sequence data by translating the data. We could start by using a very naive approach of testing each region as a possible start site. Or, given that we know the sequence of start codons, we could look for those sequences and try to test whether it looks like a true start codon sequence, or a chance event of three bases arising together.
Let's start by checking how many start codons are in the sequence by searching the region variable (a string object) for the "ATG" sequence. This could be done in a number of ways using functions of string objects.
# finds position of the first occurrence and returns it as an integer
region.index("ATG")
# split on every occurence to return a list, and count the length of the list
len(region.split("ATG"))
# best: get a list with every ATG start position
starts = [i for (i, j) in enumerate(region) if region[i: i+3] == "ATG"]
starts
OK, there are 16 occurrences of "ATG" in the genomic segment. You can see why this is not a very accurate approach to finding coding regions, since we only inserted one true "ATG" codon. Because almost any combination of three bases encodes for an amino acid, random sequences can look like a string of valid codons. Our goal is find the correct start site and read the codons in the correct phase (sets of 3) until the stop codon.
Well, another approach is that we can start from each occurrence of ATG and search by multiples of three forward to see how long until it matches the next stop codon. We might expect that non-coding regions will randomly generate a stop codon sooner. The code below will print output for each start position.
# test from each start position how many subsequent triplets are valid codons
for start_pos in starts:
# a counter for the number of codons until next stop codon
ncodons = 0
# a counter to advance 3 positions at a time
idx = start_pos
# a loop to continue until told to stop (break)
while 1:
# get codon from this position until + 3
codon = region[idx: idx + 3]
# if this is a stop codon then move on to the next start_pos
#print(idx, len(region), codon)
if GENCODE[codon] == "_":
print("start pos: {:<3}, codons until stop: {}".format(start_pos, ncodons))
break
# if not a stop codon then advance 3 more steps
else:
# advance position
idx += 3
# if next end pos is beyond the sequence end then break the loop
if idx + 3 >= len(region):
break
# otherwise record that another codon was observed
else:
ncodons += 1
Response
Yes it did, but some others had long stretches of codons as well. This can happen by chance because the three random bases can sometimes cause a start or stop codon. This means that this method can be error prone.Exons and Introns¶
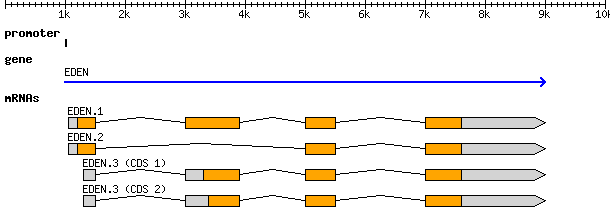
In eukaryotes the story can often be a bit more complicated. In turns out that the mRNA sequence transcribed from DNA is not always entirely translated into coding sequence, instead there are sometimes interspersed breaks of noncoding sequence that interrupt them. The coding regions are termed exons and the intervening non-coding regions introns. During transcription the region of the mRNA that is intronic is removed and the exonic regions are spliced together to form the final transcript.
Response
Introns can have stop codons in them that will not actually stop transcription, since those regions are not in the reading frame. Thus not taking to account which are regions are introns versus exons can make is confusing to identify the true reading frames.The canonical gene¶
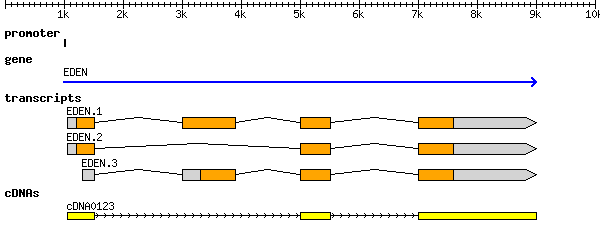
The figure below is copied from the optional reading. It shows a region of a genome that contains a gene. But how do we know this? How do we identify that one region of DNA contains a gene while another does not? Your reading for this week on A guide to eukaryotic genome assembly describes a variety of approaches.
For example, we can try to infer the location of genes from the DNA sequence itself, as we did above, but for this we generally use additional information beyond the location of methionine and stop codon sites. Instead, we'll look for longer stretches of DNA that represent binding regions for transcriptional proteins. These types of sequences are well characterized since they tend to be highly conserved (they don't accumulate mutations very rapidly and thus tend to be similar among species). The binding of transcription factor proteins to these regions affects how and when genes are transcribed. This is represented in the figure below by a "promoter" region.
Huge advances have been made in recent years in a technology called RNA-seq (see below and e'll learn much more about this in next week's reading), which allows us to identify the transcribed region through sequencing. Similarly, as was described in your first week's reading, we have long been able to decode the amino acid sequence of proteins -- in fact this was developed before our ability to sequence DNA -- and from the sequence of proteins we infer which regions have been translated or spliced from transcripts.
When all of this information is combined together we end up with a nested set of hierarchical information, where one feature (e.g., mRNA transcripts) is expected to be associated with one or more other downstream features (exons). We keep track not only of each of these features as they are identified, but also which type of evidence was used to find it in the first place. The GFF file format contains this information.
Lines of evidence¶
cDNA and RNA-seq¶
By extracting RNA from an organism, and reverse transcribing it back to DNA we create cDNA. We can then match the cDNA sequence to the genome to figure out which regions were trascribed into RNA. As you can see in the figure, cDNA can then identify the locations of transcripts. Different tissues or organs of an organism might express different genes at different times and so the cDNA from one cell will not match exactly the cDNA of another cell. We will discuss more the use of RNA-seq or exome sequencing later in the class.