Principles and Applications of Modern DNA Sequencing
EEEB GU4055
Session 12: Phylogenetics II
Today's topics
1. Review notebook assignments: Hi-C.
2. Discuss the assigned readings: Composite assembly.
3. Introduce new topic: reduced-representation sequencing.
Review of course topics
1. DNA sequencing review; and intro to Jupyter/Python.
2. Python bootcamp I: Basic objects.
3. Python bootcamp II: Scientific libraries.
4. Homology/Blast/GFF: Genome structure
5. Phylogenetics I: Sanger sequences to trees.
6. Recombination and Meiosis.
7. Inheritance and pedigrees.
8. Intro to Illumina and read mapping.
9. Intro to long-read technologies and read mapping.
10. Genome Assembly in theory.
11. Genome Assembly in practice.
12. Scaffolding.
13. Phylogenetics II: RAD-seq
14. Phylogenetics II: SNPs, gene trees and species trees
Notebook 12.1: Hi-C
Chromosome conformation capture (3C) describes the structure of the
genome within a cell; it's organization and structure. Better than
microscopy, can tell us how close together (potentially interacting)
some regions of the genome are (such as promoters and enhancers).
Hi-C: A highthroughput version of 3C is based a library preparation to
build chimeric reads followed by short-read sequencing of paired-end
reads. Creates a contact map of interactions
correlated to spatial distance.
Notebook 12.1: Hi-C
Restriction digestion; streptavidin bead extraction; paired-seq.

Notebook 12.1: Hi-C
When a genome is digested with a restriction enzyme the genome is broken into smaller fragments. Each fragment will begin and end with a characteristic overhang of the restriction enzyme. For the restriction enzyme HindIII, the recognition site is AAGCTT, and the cut occurs between the two A's in the 5' direction (A^AGCTT) such that it leaves one A at the end a fragment, and AGCTT at the beginning of the next fragment. Let's see what this looks like:
Notebook 12.1: Hi-C
def random_sequence(length):
"return a random sequence of DNA"
return "".join(np.random.choice(list("ACGT"), size=length))
def restriction_digest(sequence, recognition, cut):
"""
restriction digest a genome sequence at the given (recognition) site and
split the site at the given position (cut) to leave overhangs.
"""
# cut sequence at every occurence of recognition site
fragments = sequence.split(recognition)
# add overhang that results from sequence splitting within the recognition site
fragments = [recognition[cut:] + i + recognition[:cut] for i in fragments]
return fragments
Notebook 12.1: Hi-C
# generate a 5Mb genome
seq = random_sequence(5000000)
# digest the genome at every HindIII site
fragments = restriction_digest(seq, "AAGCTT", 1)
# print headers
print("Restriction recognition site: A^AGCTT")
print("Expected: [overhang-AGCTT][sequence][overhang-A]")
# check the beginning and end of the first 10 fragments
for i in range(10):
print(fragments[i][:5], fragments[i][5:10], '...', fragments[i][-1:])
Restriction recognition site: A^AGCTT
Expected: [overhang-AGCTT][sequence][overhang-A]
AGCTT TACAA ... A
AGCTT AATGG ... A
AGCTT CCGTT ... A
AGCTT TCCCC ... A
AGCTT AGCGA ... A
AGCTT GGATT ... A
AGCTT ATATA ... A
...Notebook 12.1: Hi-C
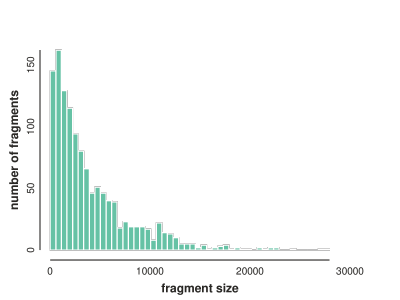
# get fragment lengths binned
flens = np.histogram([len(i) for i in fragments], bins=50)
# plot distribution of fragment lengths
toyplot.bars(
flens,
width=400,
height=300,
xlabel="fragment size",
ylabel="number of fragments",
);
Action 1: Repeat for PstI enzyme: CTGCA^G
# digest the genome at every PstI site
fragments = restriction_digest(seq, "CTGCAG", 5)
# print headers
print("Expected: [overhang-G][sequence][overhang-CTGCA]")
# check the beginning and end of the first 10 fragments
for i in range(10):
print(fragments[i][:1], '...', fragments[i][5:10], fragments[i][-5:])
#print(fragments[i][:5], fragments[i][5:10], '...', fragments[i][-1:])
Expected: [overhang-G][sequence][overhang-CTGCA]
G ... TTAGC CTGCA
G ... TTCAG CTGCA
G ... CCTTA CTGCA
G ... CTTTA CTGCA
G ... GAGCT CTGCA
G ... CCCGG CTGCA
G ... ATCAC CTGCA
G ... TATGT CTGCA
G ... AATAC CTGCA
G ... TCACG CTGCA
Notebook 12.1: Hi-C

Notebook 12.1: Scaffolding from Hi-C contact maps

Notebook 12.1: Amaranth genome

Notebook 12.1: Amaranth genome
Visit https://eaton-lab.org/data/ to view two genome reports from Dovetail Inc. assemblies.
Phylogenomics II
It's an exciting time for phylogenetics...




Phylogenomics II
Background on the method and application of RAD-seq:

Phylogenomics II
Applied phylogenomics example:
