Fundamentals of Evolution
EEEB G6110
Session 9: Phylogenetics I
Section topics
1. Homology
2. Genealogy, gene trees and species trees
3. Trees as data
4. Phylogenetics research
5. Sequence data and phylogenomics
6. Likelihood and model-based inference
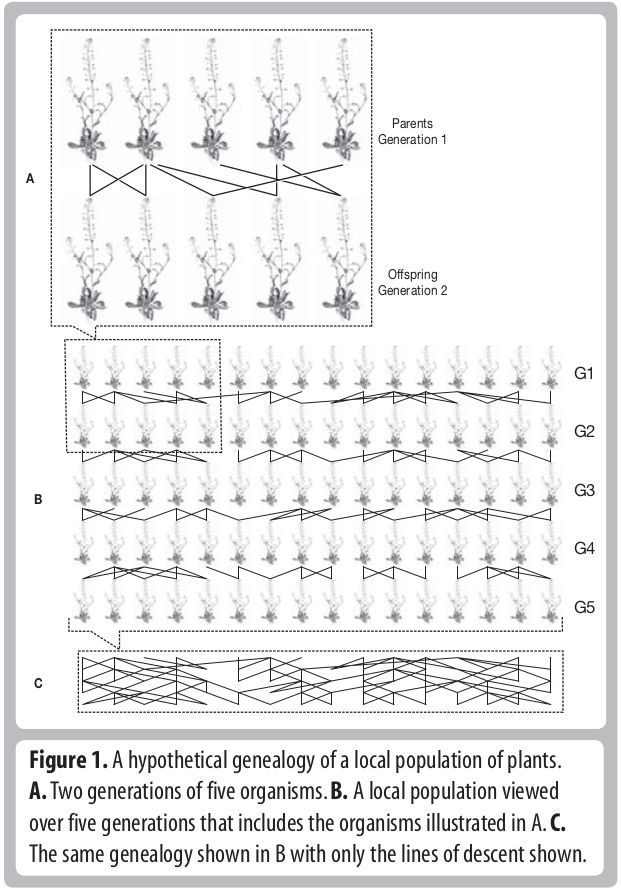
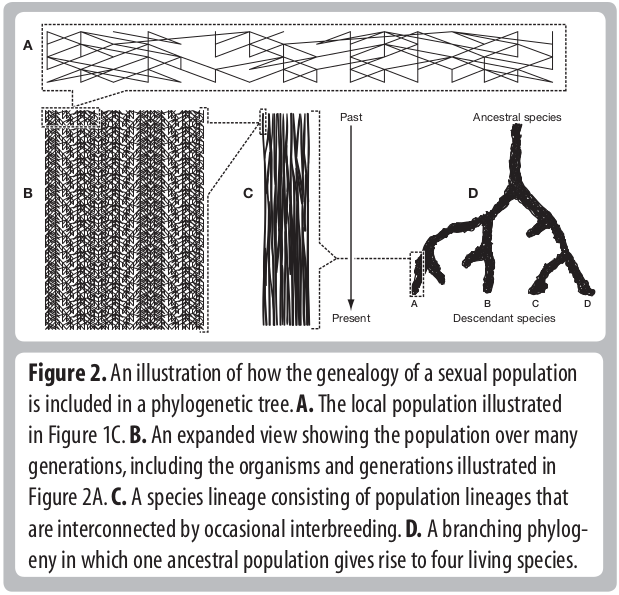
Descent and common ancestry
Homology, homoplasy and Hemiplasy
Are the characters descended from a common ancestor? Has the character state evolved multiple times? Does the evolutionary relationships of genes controlling this trait match the evolutionary relationships of the species?
Coding characters
Discrete data: DNA is discrete, there are only four possible states (A,C,G,T). But what about body mass, height, color, etc.
Characters weighting: Is one trait more informative than another? How to avoid biased sampling of characters?
Why not only use DNA?: For phylogenetics of extant taxa we mostly do. But morphology is still very relevant to phylogeny of extinct taxa (e.g., dinosaurs) and placing fossils with extant taxa
Tree Thinking
Essential to all fields of biology.
Simple but commonly done incorrectly.
Common pitfalls:
- describing taxa as being "basal".
- misinterpreting node rotations as meaningful.
- misinterpreting relationships.
- misinterpreting branch lengths.
- misinterpreting rooting of tree.
- failing to read figure captions when present.
Tree thinking exercises
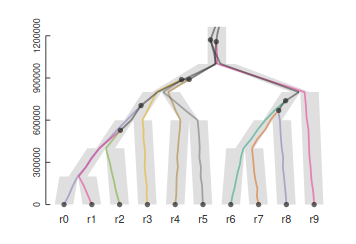
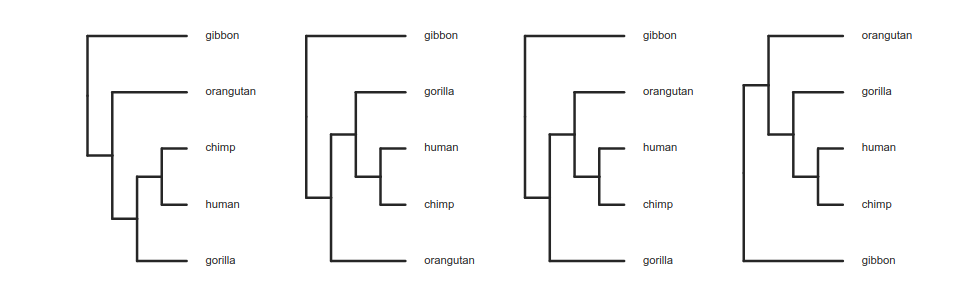
Reading trees involves interpreting the order in which lineages share common ancestors by tracing relationships backwards from the tips towards the root. Rotating nodes does not affect these relationships, even though the order of the tips changes. Which topology is different?
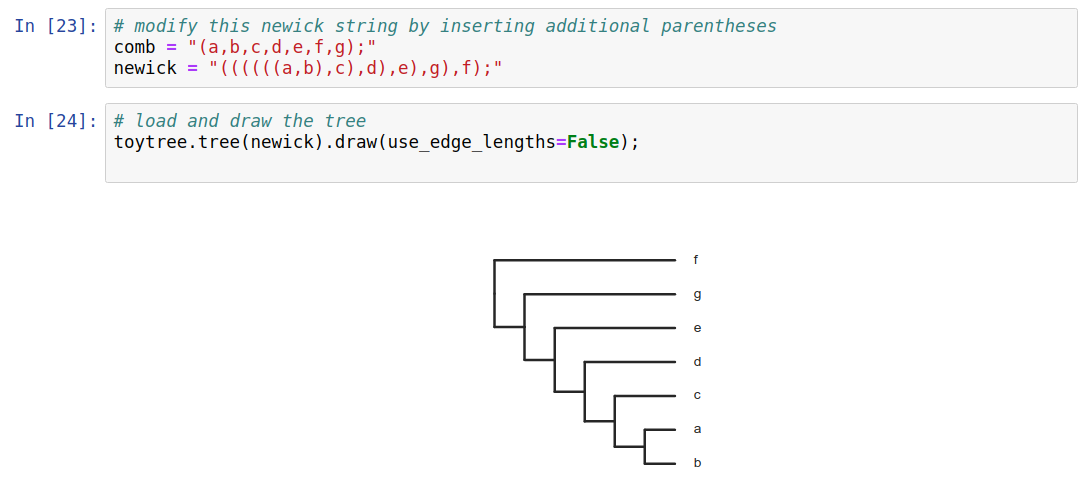
Trees as data
Phylogenetic trees are more than just pictures, they represent a data structure that can be interpreted and used in model-based analyses. Stored in Newick format.
Phylogenetic inference: examples
Methods of phylogenetic inference, and model-based historical inferences using trees, are both highly active areas of research. Many new methods are published in the journal of Systematic Biology, while countless applied examples are published in various journals, including Evolution, Molecular Biology and Evolution, Molecular Phylogenetics and Evolution, Molecular Ecology, etc.
Phylogenetic inference: methods
Collect/measure homologous characters for some number of taxa. For DNA, identifying homology typically involves targeting regions of the genome using primers, or mapping sequenced reads from the genome to the same region of a reference genome. Either way, it is based on sequence similarity. This is typically followed by a more rigorous multiple sequence alignment.

Phylogenetic inference: methods
A general outline of phylogenetic inference:
1. Propose a starting tree (e.g., random or star).
2. Score based on some criterion (e.g., parsimony, likelihood, distance).
3. Modify to propose a new tree, return to step 2.
Phylogenetic inference: Parsimony
A character matrix and a topology. Count the number of character state changes.
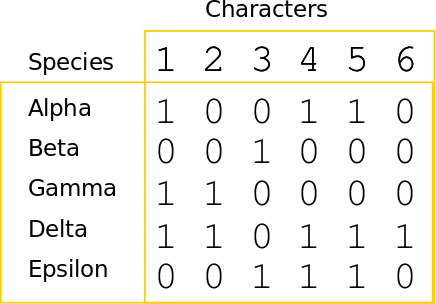
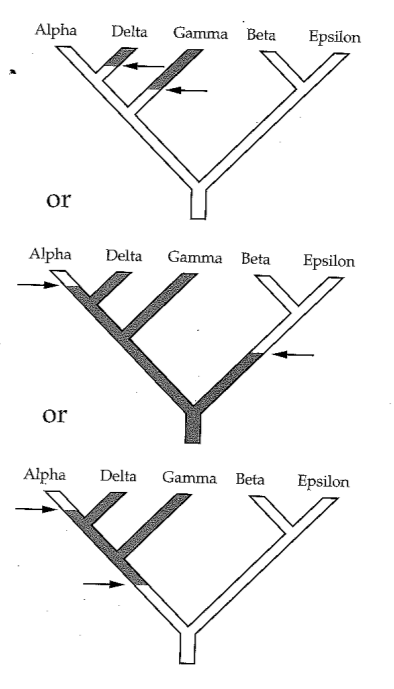
Phylogenetic inference: methods

Parsimony's pitfall
It does not account for homoplasy (repeated mutations to the same site). Or other patterns of varying rates of character change.