Skills in this course:¶
- introduction to RAD-seq assembly
- ipyrad command line (CLI)
- ipyrad Python code (API)
- introduction to jupyter
- introduction to parallel computing in Python
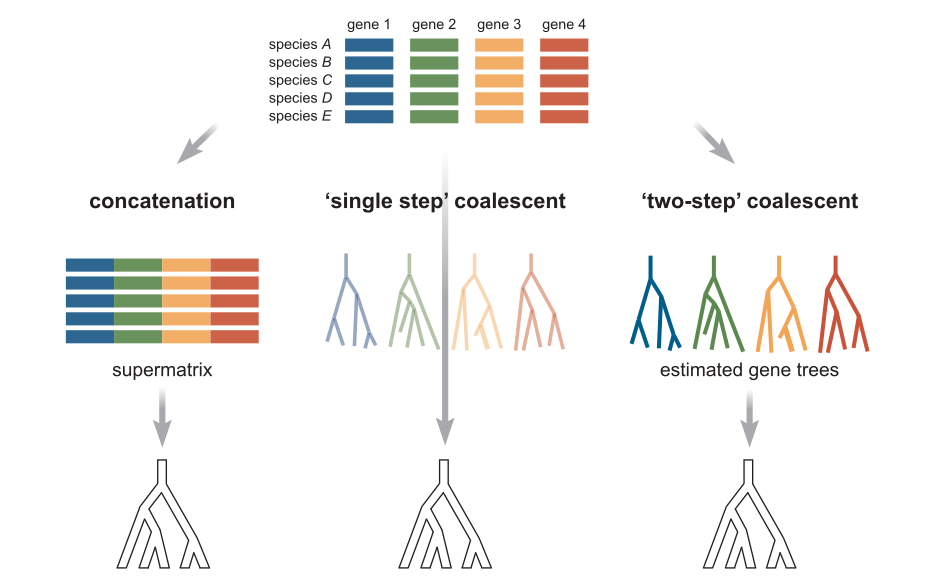
Introduction to RAD-seq assembly¶
- Short reads (usually 50-150bp) single or paired.
- Loci usually align perfectly, not tiled into contigs.
- SNP data including full sequence data.
- usually ~1e3 - 1e6 loci.
- phased SNPs within loci, not phased between loci
- anonymous (denovo) or spatial-located (reference-mapped)
Available assembly software¶
- Standard reference-mapping approaches (BWA + Picard + GATK + ...)
- STACKS
- pyRAD)
- TASSEL-UNEAK
- ipyrad
Advantages to using ipyrad over the other methods:¶
- Provides denovo, reference, and denovo-reference hybrid assembly methods
- Includes alignment steps to allow for indel variation
- Fast and massively parallelizable (hundreds/thousands of cores)
- Low memory footprint, e.g., compared to stacks.
- Branching methods support reproducibility and exploring parameter settings
- Python API supports integration with Jupyter and scripting.
ipyrad online documentation¶
The ipyrad command-line (CLI)¶
And introduction to the ipyrad setup and parameter settings.
In [3]:
%%bash
ipyrad -n tutorial
In [6]:
%%bash
cat params-tutorial.txt
In [ ]:
In [ ]:
In [ ]:
In [ ]:
In [ ]: