Phylogenomic perspectives on reproductive
isolation and introgression
Botany Conference 2019, Tucson
Deren Eaton, Columbia University
The goal of phylogenomics
Characterize evolutionary relationships from a subset of sampled genomes.

WGS vs. RAD-seq genomic sampling
Characterize whole genomes from a subset of sequenced markers.

Coalescent variation
Different genomic regions have different genealocial histories.

Can sparse SNP sampling reconstruct
genome-wide patterns?
Filtering and formatting to deal with missing data...

Viburnum Phylogeny

Viburn'ers

Viburnum global RAD sampling
From global to population-level variation.

Viburnum Orienotinus rapid radiation
~35 species from Mexico to Bolivia over ~10Ma

Viburnum Orienotinus rapid radiation
~35 species from Mexico to Bolivia over ~10Ma

Viburnum Orienotinus rapid radiation
~35 species from Mexico to Bolivia over ~10Ma

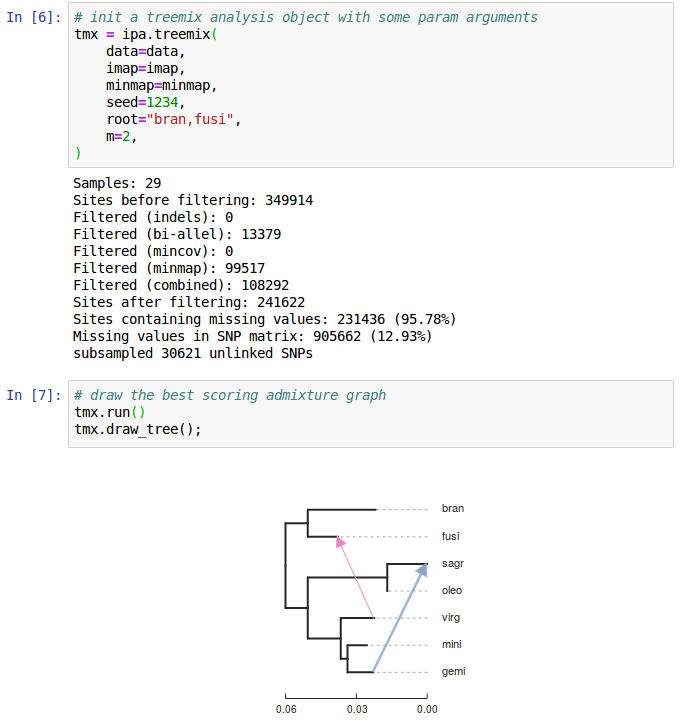
Outline: RAD-seq phylogenomics in ipyrad
1. ipyrad-analysis toolkit.
2. Gene tree extraction: concatenation.
3. Gene tree distributions: sliding window consensus.
4. Sticking with SNPs: genome-wide inference.
ipyrad-analysis toolkit (and toytree) and jupyter
ipyrad-analysis toolkit
Filter or impute missing data; easily distribute massively parallel jobs.
import ipyrad.analysis as ipa
# initiate an analysis tool with arguments
tool = ipa.pca(data=data, ...)
# run job (distribute in parallel)
tool.run()
# examine results
...
PCA: very sensitive to missing data

PCA: missing data imputed

PCA: missing data imputed

PCA + T-SNE: missing data imputed
TSNE manifold projection method (sckit-learn)

PCA + T-SNE: missing data imputed
TSNE manifold projection method (sckit-learn)

Outline: RAD-seq phylogenomics in ipyrad
1. ipyrad-analysis toolkit.
2. Gene tree extraction: concatenation.
3. Gene tree distributions: sliding window consensus.
4. Sticking with SNPs: genome-wide inference.
Missing data in phylogenetics

Missing data in phylogenetics

Missing data in phylogenetics

Window_extracter: extract, filter, format.
Reference mapped RAD loci can be "spatially binned" to form larger loci.
import ipyrad.analysis as ipa
# initiate an analysis tool with arguments
tool = ipa.window_extacter(
data=data,
scaffold_idx=0,
start=0,
end=1000000,
)
# writes a phylip file
tool.run()
Window_extracter: extract, filter, format.
Reference mapped RAD loci can be "spatially binned" to form larger loci.

Window_extracter: extract, filter, format.
Reference mapped RAD loci can be "spatially binned" to form larger loci.

Herbicide resistance among Amaranthus species.

Missing data in phylogenetics

Missing data in phylogenetics
Goal: A distribution of gene trees representing every species.
Missing data: Consensus sampling
Represent species by the consensus genotype across sampled individuals

treeslider: extract windows across chromosomes.
Runs raxml on windows and parses results into a "tree_table"
# define population groups
imap = {
"sp1": ["a0", "a1", "a2", "a3"],
"sp2": ["b0", "b1", "b2", "b3"],
"sp3": ["c0", "c1", "c2", "c3"],
"sp4": ["d0", "d1", "d2", "d3"],
}
# initiate an analysis tool with arguments
tool = ipa.treeslider(
data=data,
window_size=1e6,
slide_size=1e6,
imap=imap,
)
# distributes raxml jobs across all 1M windows in data set
tool.run()
treeslider: extract windows across chromosomes.
Consensus sampling
Recovers 5,500 informative gene trees (>50 SNPs) with no missing data across 25 taxa.
ASTRAL species tree and cloud tree of RAxML gene trees

Another data set: Quercus section Virentes
Consensus sampling yields 3X as many fully sampled loci (>30K)

Hipp et al. (2014); Eaton et al. (2015); Cavender-Bares et al. (2015)
Sliding windows
How well do concatenated RAD windows represent gene tree variation?

RAxML gene trees.
Sliding windows
How well do concatenated RAD windows represent gene tree variation?

Astral species trees inferred from gene trees.
Clade weights (sensu Martin et al. 2017)
Chrom 1 weighted support for a (Cuba, Florida) vs (Cuba, Mexico)

Missing data in phylogenetics

SNP-based species trees
SNAPP: joint inference of gene trees and species trees.
(Bryant et al. 2012)
SVDquartets: infers quartet trees from SNPs and joins these into a species tree.
(Chifman and Kubatko 2014)
Advantages to SVDquartets-based methods
Each quartet is inferred independently: missing data has almost no effect.
Huge sptrees: 204 Viburnum taxa; 2.4M SNPs; 176K SNPs/quartet; >70M quartets.
It is fast! tetrad: 40 cores, one bootstrap ~24 hours.

Conclusions
1. With ipyrad-analysis it is easy to run dozens of analyses optimized for RAD missing-ness with a few lines of code.
2. Concatenating RAD loci in scaffold windows, and consensus or imputation sampling, dramatically improve the utility of RAD.
3. SNP based methods are in their infancy, but work well with RAD data.
Announcement
RADcamp wetlab AND bioinformatics workshop in New York City Oct. 2019
Bring your DNA samples.
Library preparation and sequencing will be free.
(sponsored by SSB, SSE, Columbia, CCNY).
https://radcamp.github.io/NYC2019/
Acknowledgements
Viburn'ers: Donoghue-lab, Edwards-lab, M. Olson, I. Cacho.
ipyrad development: Isaac Overcast
Eaton lab members
Funding: NSF DEB 1557059; Columbia University