Jekyll2024-03-12T12:35:46+00:00https://eaton-lab.org/feed.xmlThe Eaton Lab at Columbia UniversityBotany / Phylogenetics / BioinformaticsLatex guide2020-08-18T00:00:00+00:002020-08-18T00:00:00+00:00https://eaton-lab.org/articles/latex-guideWhat is latex, and why use it?
Latex is a coding language for formatting text into a typeset document, such
as a PDF. It is used widely in scientific writing and publishing as a way of
reusing a design template so that you as the writer can focus on content.
I prefer writing in Latex because it allows me to use coding practices like
git version control, commenting sections of text, and writing
text in a fast and responsive text editor. Most importantly, it also
makes working with bibtex citations super easy and convenient. Because latex
works seamlessly with git and GitHub it helps me to stay organized by
creating GitHub repositories for each manuscript, so that me
and my collaborators can all work on a cloud-based document together.
Installing latex
My instructions below are for Ubuntu Linux, and will work in the Windows
subsystem for Linux as well. If you are on MacOS I’m sure you can find
similar installation instructions using google. In your linux bash terminal
use apt to install latex with the following command, which may take a
few minutes to install.
sudo apt install texlive-latex-extra
That’s it, you are now ready to make a .tex document and compile it with
latex. In particular, we will use the program pdflatex to compile tex
files into PDF documents.
Compile your first tex file.
Open any text editor and create a new file called hello-world.tex and
write into it the text below and save the file.
\documentclass{article}\begin{document}
Hello world
\end{document}
This is a super simple tex document that includes the very minimum that is
required to compile. The same tex commands are copied below but with additional
notes for each section added by using the comment character (%). The ability
to add comments to your tex files is a really important and powerful component
to writing in latex. Commented lines are not compiled into the PDF. They can
be left in the tex file as notes to yourself or collaborators, or to save
earlier versions of a text while you are in the process of editing.
% This is a comment.% The document class sets an overall style for the document % and is usually the first command in a tex file.\documentclass{article}% additional add-on packages can be loaded here (see later examples)% or additional styling options are added here, before beginning the doc.% This command starts the document. Everything after this is intended to % be printed into the document (except comments). Everything% before this involves loading styles and options that will be used in % this section to style text and images.\begin{document}% This text is part of the document. This comment line however will % not appear in the document.
Hello world
% This ends the document. Anything after this will be ignored.\end{document}
You can now compile the tex document (using either one of the two tex files
above since they are identical other than comments) by calling the command below
from your bash terminal. Make sure to reference the full or relative path to
the tex file that you just created. This will print some information to the
terminal about what it is doing and any errors it encountered. The output
is mostly mumbo jumbo. After it finishes use ls to look in your current
directory. You should see a new hello-world.pdf file containing the typeset
document. A few additional files will also be created which contain errors
or auxiliary information such as citations. You can generally ignore those
other files.
pdflatex hello-world.tex
Setup latex with your text editor (e.g., sublime)
Now that you’ve compiled a tex file from your bash terminal, you can move
to a more advanced setup, which involves compiling the tex file directly
from your text editor, thus avoiding the additional step of having to
open a terminal. There are several options for this, including many
dedicated latex editors/IDEs that are designed specifically to display
a PDF next to your tex document (and online versions of this like overleaf).
These can be nice, but I find them overall to be kind of clunky and ugly.
Instead I recommend learning to use latex in a powerful coding text editor
such as sublimetext or vscode. This allows you to learn and use the same
set of hotkeys and keystrokes to write text efficiently and maneuver around
lines and paragraphs that you use when writing code.
For sublime text you can find instructions online for how to set it up for
latex. For me, this involved adding a latex command to the build system to
compile a .tex file when I press the F7 key. This makes it very easy to edit
the .tex in my editor, press F7, and see the changes in the PDF document.
This can also be setup on Windows as well, where latex is installed in
your Linux subsystem, but Sublime is installed in Windows,
instructions here.
Using comments
A benefit of using latex for writing large documents is that you can very
easily comment out regions of the text that you wish to change, leaving behind
a copy of the unedited version. I use this feature a lot when writing or
editing. Unlike word or googledocs you don’t need to worry about whether your
edits are easy to read over the previous version, and making lots of edits
will not lag the system. I leave a copy of the unedited version in a comment
until I’m satisfied it is no longer needed and then remove it.
Writing for version control
I recommend reading a simple latex tutorial to learn how the latex
syntax works. For example, similar to markdown, line breaks are ignored
in latex. This is a useful feature for interpreting your text like code.
I always manually break paragraphs into lines that <=80 characters to avoid
line wrapping when working in latex. This makes it so that when you push
or pull changes with git the changes to each line will be highlighted and
easy to find. If you write paragraphs as a single unbroken line that is
wrapped by your editor then you will not be able to find changes in each
version as easily.
Pushing changes to GitHub
Push changes to git frequently, especially when working with collaborators
to avoid conflicts from arising when you both edit the same text. If
conflicts do arise, use your text editor to go through one by one the regions
between the >>> and <<< markers to select which version of the text
you wish to keep. Then delete the delimiter markers.
Only commit and push the .tex file to git, not the PDF or auxiliary
files. You and your collaborators can each compile the PDF anew when
you load the changes to the .tex file. Git is great for versioning
text-based documents like .tex but not PDFs. Add, commit and push
new changes like below.
# pull in changes from your collaborators
git pull
# example git commit to push changes to a manuscript
git add hello-world.tex
git commit -m"added genomics section to Methods"
git push
Conda is a work in progress, and the best practices evolve quickly. This is
my current recommended best practice, aimed at avoiding conflicts among
packages, and preventing the need for total reinstallations.
Fresh installation
Download the latest Miniconda3 and install into your home directory.
# install in batch mode
bash Miniconda3-latest-Linux-x86_64 -b
If conda is not yet in your path (e.g., this is your first time installing)
then add it your path by calling:
~/miniconda3/condabin/conda init
Create a working environment
It is best not to install additional packages into your base environment.
Instead, create one or more environments. I’ll create an environment using
Python 3.7 since 3.8 is not yet widely supported.
conda create -n py37 Python=3.7
conda activate py37
Add conda-forge as your constant default channel
Remember that the order in which you list channels is important, since the
they are checked in order to choose priority. It is best to install all packages from the same channel as much as possible to reduce conflicts.
Conda-forge is the most expansive channel that also has the latest updates.
Even if you need to install a package that is on bioconda, it is best to list
conda-forge before bioconda so that any dependencies of the bioconda package will be pulled in from conda-forge.
When you are doing development you often want to install software locally
with pip so that you can incorporate changes in your code instantly into your
development environment. I recommend doing this with the option --no-deps like below to ensure you do not accidentally install dependencies with pip,
since this can cause conflict problems. Here is an example with ipyrad
cloned from github.
# install ipyrad from conda to get all dependencies
conda install ipyrad -c conda-forge -c bioconda
# clone the ipyrad repo to get git development version
git clone https://github.com/dereneaton/ipyrad.git
# cd into the repocd ipyrad/
# do local pip install (-e) with --no-deps
pip install-e.--no-deps
]]>Deren Eatonde2356@columbia.eduEaton lab server guide2020-04-19T00:00:00+00:002020-04-19T00:00:00+00:00https://eaton-lab.org/articles/pinky-loginConnecting to pinky
This guide will walk you through the recommended steps to get set up
for using the pinky server and for following shared use best practices.
1. request access
Write to Deren to request a username and password to be setup
for you on pinky.
2. Create a GitHub account
If you don’t yet have one, create an account.
3. generate a public SSH key
On your laptop run the command below to generate a private and public key
pair. This will request that you enter a passphrase, if you want you can just
hit enter to leave the passphrase blank. This will generate two files placed
in your ~/.ssh folder. The private key stays on your laptop and the public key will be sent to pinky so that the two files can be matched up
when you try to connect.
ssh-keygen -t rsa -b 4096 -C"deren@sacra"
4. upload your public SSH key to your GitHub account
Your public key can be shared publicly, and used for a variety of security
purposes. To ensure that you do not lose it I recommend uploading it to
your GitHub account. Follow the instructions here: https://jdblischak.github.io/2014-09-18-chicago/novice/git/05-sshkeys.html.
Once your key is uploaded send Deren an email with your GitHub
username and he will pull your public key onto pinky so that you
will be able to login.
5. setup your laptop for easy ssh login
Next edit your SSH config file on your laptop to create a shortcut name to
reference the pinky server. This makes it so that you do not need to write
out the full IP address and username when you login. Replace the {username}
with your own name in lower case (e.g., deren) without brackets.
# nickname the server pinky (ENTER YOUR USERNAME w/o brackets)touch ~/.ssh/config
echo-e"
Host pinky
Hostname 128.59.23.200
User {username}
"> ~/.ssh/config
Finally, you can now login to pinky from your terminal by just typing:
ssh pinky
]]>Deren Eatonde2356@columbia.eduAssembling a plant genome on google cloud2019-11-24T00:00:00+00:002019-11-24T00:00:00+00:00https://eaton-lab.org/articles/gcloud-projectAssembling a plant genome with nanopore data
Our goal is to assemble a genome for the flowering plant Pedicularis cranolopha. We originally estimated the genome size to be 1-2Gb and generated ~180Gb of Illumina PE 150bp reads, 250Gb of nanopore reads (avg. read len ~30Kb) and ~350Gb of PE 150bp Illumina Hi-C data in addition to XXGb of RNA-seq data for genome annotation. Here I will focus on how we setup a google cloud instance for genome assembly using canu and shasta.
Setup a google cloud computing project
Log into https://console.cloud.google.com/ to create a free account (you may be eligible for free academic credits). Then create a new project. Ours is called “liuliu”. My postdoc Jianjun Jin who is leading the bioinformatics for this project also created a personal account and I added him under the IAM section as a project “owner” to have full permissions.
Create a storage bucket
gs storage buckets are convenient for storing data long term as well as for transfering files between different locations. I backed up all of our data onto a bucket which takes up about 500Gb of space. You can create a bucket from the dropdown toolbar in the upper left corner: find “storage”, then “storage” again to open the bucket storage page. There you can create a new bucket or modify existing buckets to set access rights. The bucket with our genome data is called “liuliu”. This is where we will store the raw data.
Setup gcloud and gsutil to transfer data to gcloud
I followed instructions from here to install and setup the gsutil tool on my local computer where the raw data is saved: https://cloud.google.com/storage/docs/gsutil_install#linux. The init command allows you to securely connect to gcloud using google authenticator in your browser.
# Enter the following at a command prompt:
curl https://sdk.cloud.google.com | bash
# Restart your shell:exec-l$SHELL# Run gcloud init to initialize the gcloud environment:
gcloud init
Copy data to the bucket
Once you are logged in you can see the available buckets visible to your account using the following command on your local machine:
gsutil ls-l
And then transfer local files to the cloud bucket using the cp command on your local machine:
gsutil cp file.txt gs://liuliu
Create a hard disk on gcloud
A persistent disk can be used like a scratch drive on an HPC system to store processed data such as temporary files created during the genome assembly. According to the canu documentation you should have ~3 Tb of free disk space for a mammal or human-sized genome, but up to 10-20Tb for a highly repetitive genome such as a plant. Our genome size is estimated to be smaller than human (~1G) and not particularly repetitive (~2%) based on kmer statistics, so I created a 6Tb disk to be safe. When finished with the assembly we will transfer the long-term data files back to the storage bucket and delete disk. The disk was created by selecting from the toolbar “Compute Engine” and then “disks” and I named it “scratch”.
Starting an instance and format the scratch disk
I created an instance (named assembly) in project liuliu that boots an Ubuntu 19.04 from a 10Gb disk and has the 6Tb ‘scratch’ disk attached containing the raw data. The instance (for now) is 32vCPUs and 120Gb of disk. This seems like a reasonable amount of resources for our initial analyses with canu, which requires only about 16Gb per node. We will want more RAM for later shasta assembly, and we can stop and edit the instance at any time later to change the resources.
Once the instance has started I then connect to it with SSH. I followed instructions to format and mount the scratch disk on the compute instance here.
# on the assembly instancesudo mkfs.ext4 -m 0 -F-Elazy_itable_init=0,lazy_journal_init=0,discard /dev/sdb
sudo mkdir-p /scratch
sudo mkdir-p /scratch
sudo mount -o discard,defaults /dev/sdb /scratch/
sudo chmod a+w /scratch
# set to re-attach on restart of instancesudo cp /etc/fstab /etc/fstab.backup
echo UUID=`sudo blkid -s UUID -o value /dev/sdb` /scratch/ ext4 discard,defaults,nofail 0 2 | sudo tee-a /etc/fstab
Transfer raw data to the scratch disk
Copied from the bucket to the scratch dir for faster i/o access.
To install canu’s dependencies and ensure binaries are accessible to all users I installed canu into the /opt/conda/bin directory.
# install conda in /opt/ so it is available to all userscd
sudo bash Miniconda3-latest-Linux-x86_64 -p /opt/conda -b# activate conda in path so that dependencies are found (e.g., java).source /opt/conda/bin/activate
conda init
exec-l$SHELL# install with condasudo conda install canu=1.9 -c bioconda -c conda-forge
Clean and trim nanopore reads with canu
This is what we plan to run first (do we need to run the correct and trim steps multiple times?). Then we will probably try a fast shasta assembly of the trimmed and cleaned reads. Then if that goes well we will start a canu assembly as well. The shasta assembly will probably require changing the instance to a high mem node.
For high coverage data this makes it faster:
correctedErrorRate=0.12
Discard short reads (default=1000).
minReadLength=10000
Don’t look for overlaps shorter than 500bp (default=500)
minOverlapLength=500bp
IN PROGRESS …
Install shasta from source
For best performance build it on the machine (instance) that we plan to use for the assembly (high memory node instance). This takes about 10 minutes to
install: https://chanzuckerberg.github.io/shasta/BuildingFromSource.html
Or, to build a version that is transferrable between machines add the following flag to the cmake call: -DBUILD_NATIVE=OFF.
]]>Deren Eatonde2356@columbia.eduLogin to HPC passwordless2019-11-19T00:00:00+00:002019-11-19T00:00:00+00:00https://eaton-lab.org/articles/login-to-HPC-easilySet up a shortcut to login by SSH
In a linux or OSX terminal you will have a hidden directory in HOME called ~.ssh
which contains files for setting preferences or login credentials to make it simpler
and faster to login to remote systems. Let’s start by setting a shortcut for
the two clusters at Columbia in a file called ~.ssh/config. The code below
shows the typical longform SSH login command and the shorter version that we will
be able to use once you setup your config file.
# what you do now
ssh username@habanero.rcs.columbia.edu
# what you want to be able to do
ssh habanero
To setup the config file use a text editor like nano to create and edit the config
file by calling nano ~/.ssh/config and then enter the following being sure to
replace USERNAME with your actual username.
Host habanero
Hostname habanero.rcs.columbia.edu
User USERNAME
Host moto
Hostname moto.rcs.columbia.edu
User USERNAME
Setup passwordless login
Great, now that we can call the command to login to the cluster more easily let’s
also make it so that you do not need to enter a password. We can do this by sharing
SSH credentials between your laptop and the cluster. This is a two-step process.
1. Generate an SSH key
Enter your email address here of course. This will prompt you to enter a password
for which you should enter the password you wish to use to login to the cluster
(you can set this to not ask later).
ssh-keygen -t rsa -b 4096 -C"user@email.org"
2. Send SSH key to the HPC
Now we send the key to the cluster.
ssh-copy-id -i ~/.ssh/id_rsa.pub habanero
And repeat for the other cluster.
ssh-copy-id -i ~/.ssh/id_rsa.pub moto
That’s it. You should now be able to login more efficiently.
The Eaton lab is at Evolution 2019. Pictured from left to right are
graduate students Patrick McKenzie and Jared Meek, then myself, and postdoc
Sandra Hoffberg. If you see us at the conference come say hi.
Talk slides
In case you missed my talk, or saw it and want to revisit the slides, you can
access an online version here of the slides here.
]]>Deren Eatonde2356@columbia.eduHosting a jupyterhub server on a static website2019-06-24T00:00:00+00:002019-06-24T00:00:00+00:00https://eaton-lab.org/articles/setup-jupyterhubThe problem
As a biologist and instructor working in computational genomics I frequently
teach workshops and classes aimed at introducing new computational methods
that draw on a variety of computer languages and software. And, as anyone who
teaches computational methods knows well, the most difficult part of running
a workshop is troubleshooting installation problems on the varied computers of participants (that one person with a chromebook, or Windows 95);
a task that feels particularly thankless when only teaching a one-time workshop. An alternative strategy that would allow participants to jump straight into learning code on a system with all requirements pre-installed can save a ton of time.
A convenient solution
I love teaching with jupyter notebooks and so I will focus on setting up an environment that easily allows users to connect to pre-loaded tutorial notebooks, and to modify their environment (e.g., install more software) if needed. After trying many different solutions (HPC accounts, collaboratory, binder), I’ve settled on hosting a jupyterhub from my lab workstation as my favorite option. This post describes how I set this up and why I think it’s
great. I provide instructions so others can replicate or improve this setup, and to document the steps involved (so I can remember them!).
Jupyterhub
My jupyterhub server is accessible from my lab website
(https://eaton-lab.org) on a subdomain
(https://jhub.eaton-lab.org). The site itself
is a simple static site hosted on github, which anyone can set up for free,
and the domain name costs $12/year. No matter what computer you are using (even a phone) you can connect and login to this URL and connect directly to a jupyter notebook. The GIF below demonstrates the general idea:
The key steps involved are (1) deciding how to
authenticate users (e.g., passwords versus external authenticators like
Google or GitHub); (2) setting up SSL so that authentication data is encrypted; and (3) setting up user accounts. The latter task can be done in a number of ways: for example, you can make separate accounts for each user on the workstation/computer running the server, or, you can sandbox users inside docker containers (or kubernetes, or similar alternatives).
I set up Docker containers as I found it was the easiest solution to allow both long-term users (e.g., lab members) and temporary users (e.g., workshop
participants) to both use the system safely. Docker also allows me to provide
pre-installed software, while also allowing users to install additional
software into their own isolated and persistent containers. Finally, for
temporary users, I can easily clean up and remove their containers when the
workshop or class is finished.
jupyterhub requirements
My setup is based on the zero to jupyterhub tutorial,
but I deviated from these instructions a bit as well. For me, it was important
to set up the software environment how I wanted it, and to host the server
locally, not on a paid amazon server or the like. I’ve
tried to distil the instructions from there to further explain the sections
that I found most confusing given my limited experience with networking. Here
is what we will need to get started:
A linux/unix system -- (in my case Ubuntu.)
Python 3.4 or greater -- (we'll install a Py37 conda env.)
A static IP address -- (easy to get.)
TLS certificate and key (easy to get.)
Domain name (purchase, get from your institution, or use free options.)
Step 1. Get a static IP address for your server
If you are at a University you can ask your IT department to set up a static
IP address for you. They will send it to you in an email. Otherwise
google “how to get a static IP”. It is the IP address you will run your server
from.
Step 2. Register a domain and subdomain
My lab website is hosted by GitHub using their free service for hosting static
sites. These are easy to set up by placing a bit of code into a GitHub
repository. In order to link this site (eaton-lab.github.io) to
a jupythub server, however, I needed a domain name that I could control.
So I purchased the domain eaton-lab.org from google domains ($12/year),
and set it up to forward my GitHub site to the new domain. I explain below
how to do this. If you’re on a tight budget there are free services for
getting a domain (https://www.noip.com/),
which worked for me just fine when I was first testing this out.
To set up domain forwarding for a GitHub site go to settings in the GitHub
repository for your site and set the “custom domain” to your new domain name.
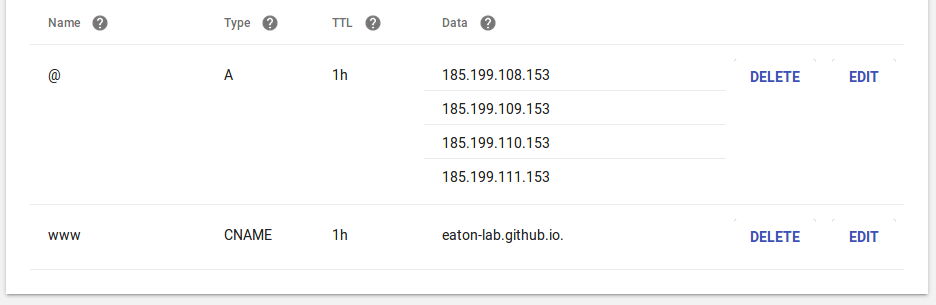
Then go to the DNS settings on domains.google.com, or where ever your domain
is hosted, and enter the GitHub IP address as the A record like below (just
enter the same values I did below), and then enter the GitHub domain address
as the CNAME record.
Your GitHub site will now be served on the domain name that your purchased
(it takes a few minutes to sync). I then set up my jupyterhub server to be
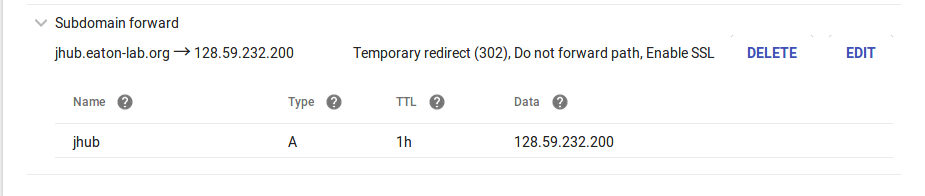
accessible from this site on a subdomain (jhub.eaton-lab.org) by
registering a subdomain like below.
This is where you need to enter the static IP address. When we start
the server we will tell it to serve at that IP address.
Step 3. Install miniconda3 in /opt/
The easiest way to get all required software is to use conda. We will need
to be able to run jupyterhub using sudo, and in a place that is accessible to all users (e.g., not from your user home directory), and so it’s easiest to install a separate and dedicated conda dir just for running your jupyterhub.
A common place for this is in /opt/. The commands below will install a fresh
miniconda installation into /opt.
## download a new miniconda3 installer (if on Mac use the Mac version!)
curl -O https://repo.continuum.io/miniconda/Miniconda3-latest-Linux-x86_64.sh
## install miniconda dir into /opt/miniconda## -b agrees to the license terms## -p sets the install prefix pathsudo bash Miniconda3-latest-Linux-x86_64.sh -b-p /opt/miniconda
Step 4. Install jupyterhub in /opt/miniconda/bin/
Now install jupyterhub and a few additional dependencies. Again, we’ll use
sudo when installing the software, and because the user environment is hidden
when using sudo, you need to write out the full path to the conda or pip
binary.
## write the full path to the opt/ conda binarysudo /opt/miniconda/bin/conda install-c conda-forge jupyterhub notebook ipykernel
And then install a few extra tools with pip, which we’ll be using to
set up a user authenticator, and to run docker.
## write the full path to the opt/ pip binarysudo /opt/miniconda/bin/pip install oauthenticator dockerspawner netifaces
Step 5. Create jupyterhub directory in /srv/
Servers facing out to the world should be run from the /srv/ directory,
which also requires sudo permissions to modify, so let’s start by
creating a directory for our config files there. This directory will
contain some sensitive information, so for some types of setups
you may want to modify the steps here to ensure users cannot
see the information which could provide them access to your system.
If you are following the same setup as me then connected users will end up
in isolated Docker containers when they login, and so they’ll never have
access to this location and security is not an issue.
## create a new server directory and cd into itsudo mkdir-p /srv/jupyterhub
## set permissions so users can rwx heresudo chmod ugo+rw /srv/jupyterhub
Step 6. Generate SSL certificates for your domain
We need to generate the SSL certificate that will allow users to “trust” our
site when they connect to it. Since we have a domain name registered, we can
generate a cert and key file using the free tool certbot. You can get
instructions for installing certbot on your system at
https://certbot.eff.org. I copied the Ubuntu
instructions below.
To generate the certificates call the certbot program and provide it your
domain name. This will generate a 90 day certificate and a job to renew the
certificate every 30 days. The files will be written to
/etc/letsencrypt/live/[domain name], which you will need to use sudo to
look at.
# generate certificatesudo certbot certonly --standalone-d jhub.eaton-lab.org
# test out the renewal processsudo certbot -renew--dry-run
...
IMPORTANT NOTES:
- Congratulations! Your certificate and chain have been saved at:
/etc/letsencrypt/live/jhub.eaton-lab.org/fullchain.pem
Your key file has been saved at:
/etc/letsencrypt/live/jhub.eaton-lab.org/privkey.pem
...
Step 7. Start to configure Jupyterhub
The jupyterhub config file can be intimidating when you first look at it
because there so many lines of options. But most of those lines
are commented out by default, meaning that they have not effect – the file
doesn’t actually do anything until you edit it. Generate the config file like
below. Then we will edit it by adding the basic information required to
securely connect to the jupyterhub.
## cd into jupyterhub dircd /srv/jupyterhub
## generate config file
/opt/miniconda/bin/jupyterhub --generate-config
## generate a random cookie secret and store it in /srv/jupyterhub
openssl rand -hex 32 > /srv/jupyterhub/jupyterhub_cookie_secret
%% editing /srv/jupyterhub/jupyter_config.py
## Configuration object for jupyterhub
c = get_config()## SSL connection
c.Jupyterhub.jupyterhub_cookie_secret ="./jupyterhub_cookie_secret"
c.JupyterHub.ssl_key ="/etc/letsencrypt/live/jhub.eaton-lab.org/privkey.pem"
c.JupyterHub.ssl_cert ="/etc/letsencrypt/live/jhub.eaton-lab.org/fullchain.pem"
c.JupyterHub.port = 443 # standard port for SSL connections
c.JupyterHub.ip ='128.59.232.200'# enter your static IP here
Step 8. Configure an Authenticator
We now want to add a method for authenticating usernames and passwords
so that users can log into our system and trust that we are not stealing
their information. One easy way to do this is to use external authenticators
serviced by GitHub or Google. I use GitHub. To do this we need to first
create an OAuthenticator app on GitHub. Login to your GitHub account and go
to Settings by clicking on your icon in the upper right corner, and then you
should see a list of tabs on the left side of the next screen. Choose
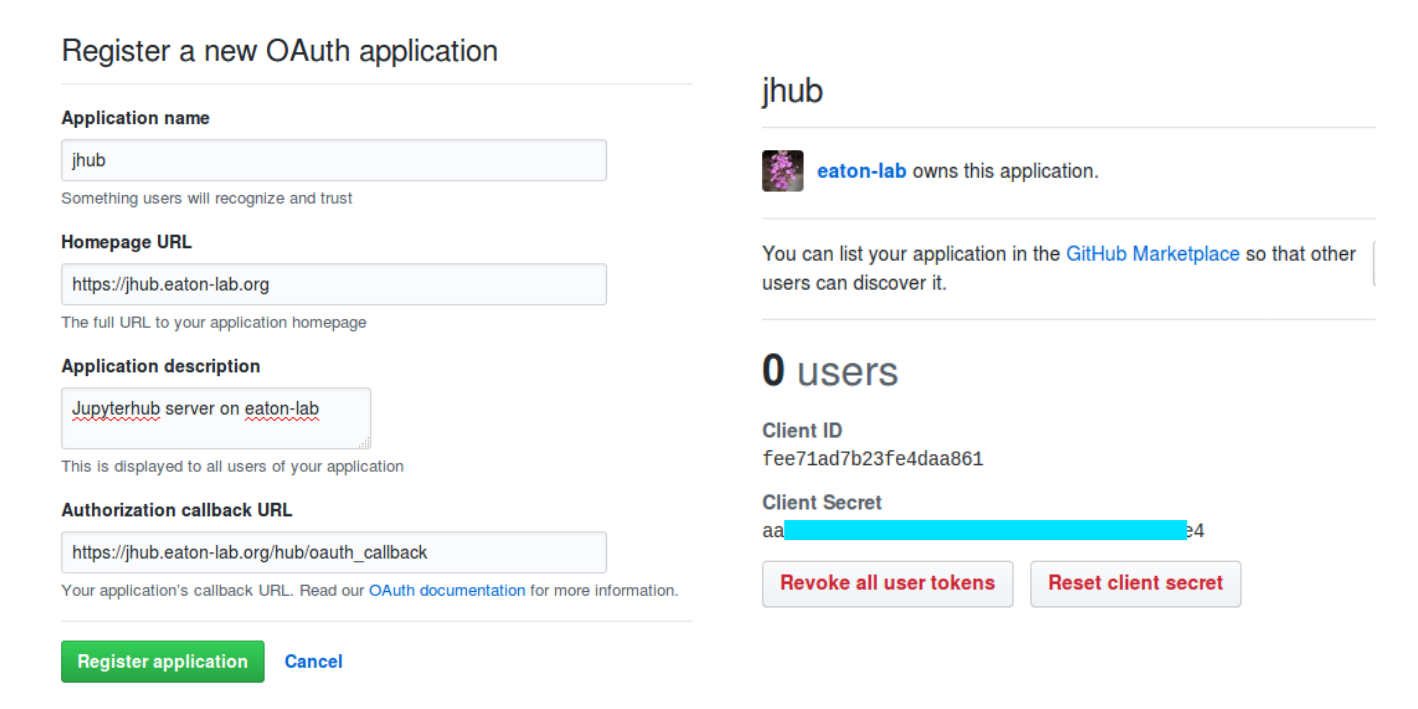
Developer settings, and then click on a button that says New OAuth App.
Register your app by giving it a name, we then need to save the callback URL.
This will generate a Client ID and Client Secret. We will need those keys.
Let’s then store these values in the jupyterhub config file after telling it
that we are using the GitHubOAuthenticator object as our authenticator.
The oauth callback url, client_id, and client_secret can be found on your
GitHub app (see above). Note: do not share your client_secret.
%% editing /srv/jupyterhub/jupyter_config.py
## Add authentication through github
c.JupyterHub.authenticator_class ="oauthenticator.GitHubOAuthenticator"
c.GitHubOAuthenticator.oauth_callback_url ='https://jhub.eaton-lab.org/hub/oauth_callback'
c.GitHubOAuthenticator.client_id ='fee71ad7b23fe4daa861'
c.GitHubOAuthenticator.client_secret ={hidden}## you would copy the real secret here
Now we need to tell jupyterhub which GitHub usernames are approved to login
to our server (when running a workshop or class you can also add these on the
fly later). I add my own username as an administrator, and optionally you can
add a usermap dictionary that will translate GitHub login names to the user
names on the workstation if they are different.
%% editing /srv/jupyterhub/jupyter_config.py
## Who is allowed access the server
c.Authenticator.admin_users ={"eaton-lab"}
c.Authenticator.whitelist ={"eaton-lab",
"isaacovercast",
"pmckenz1",
"camayal",
}
c.Authenticator.username_map ={"eaton-lab": "deren",
"pmckenz1": "patrick",
"isaacovercast": "isaac",
"camayal": "carlos",
}
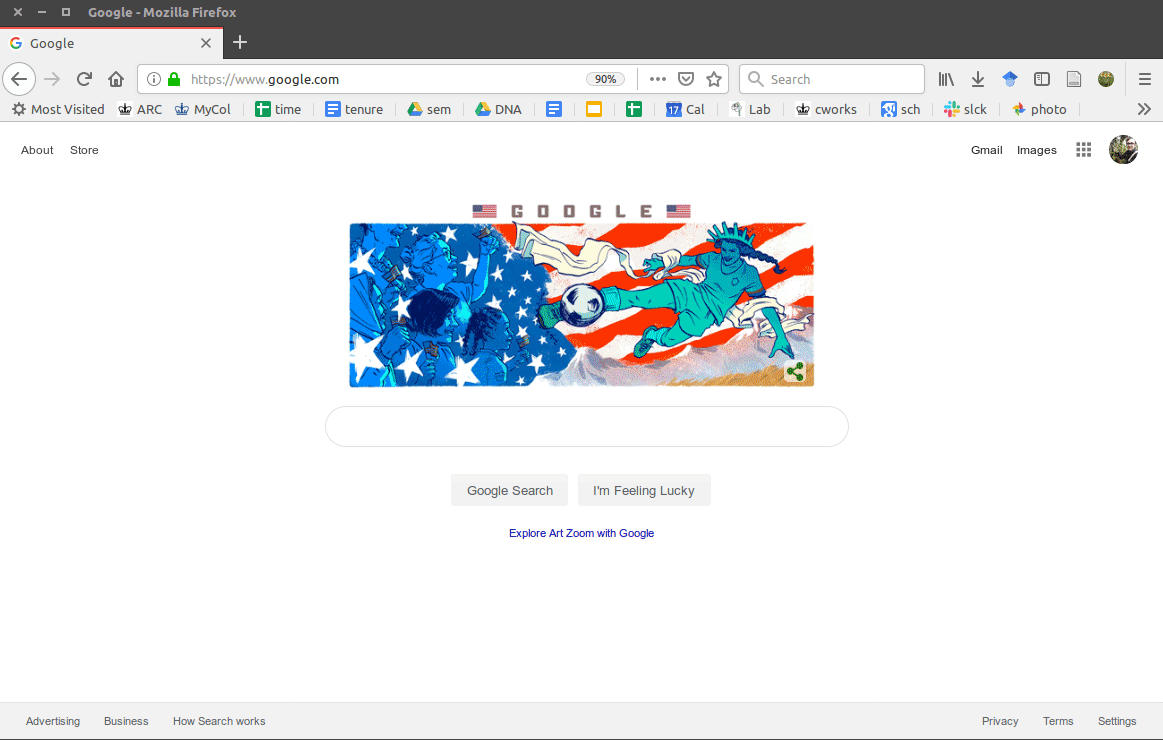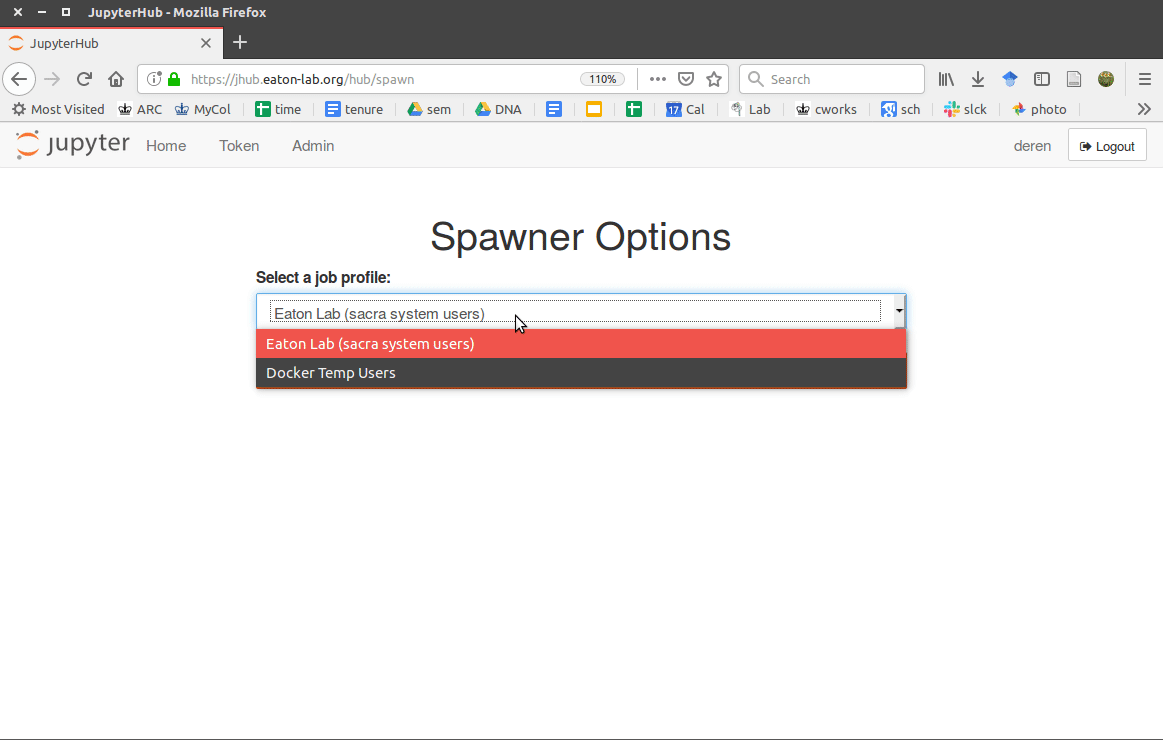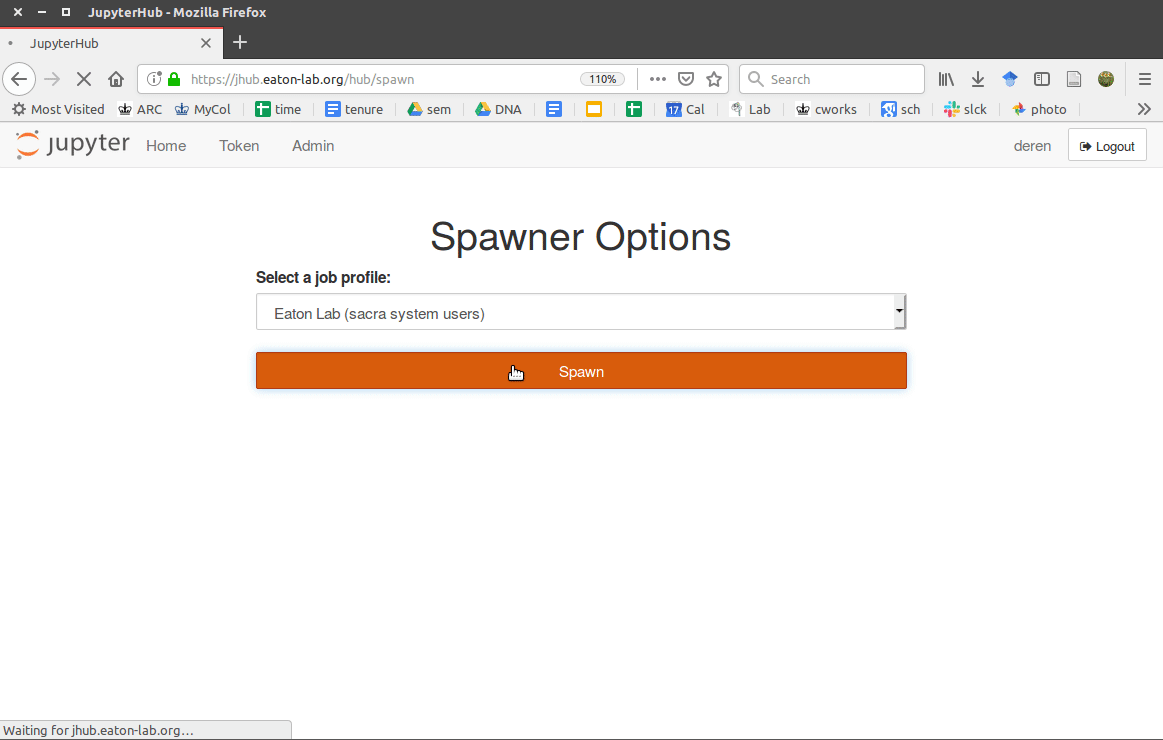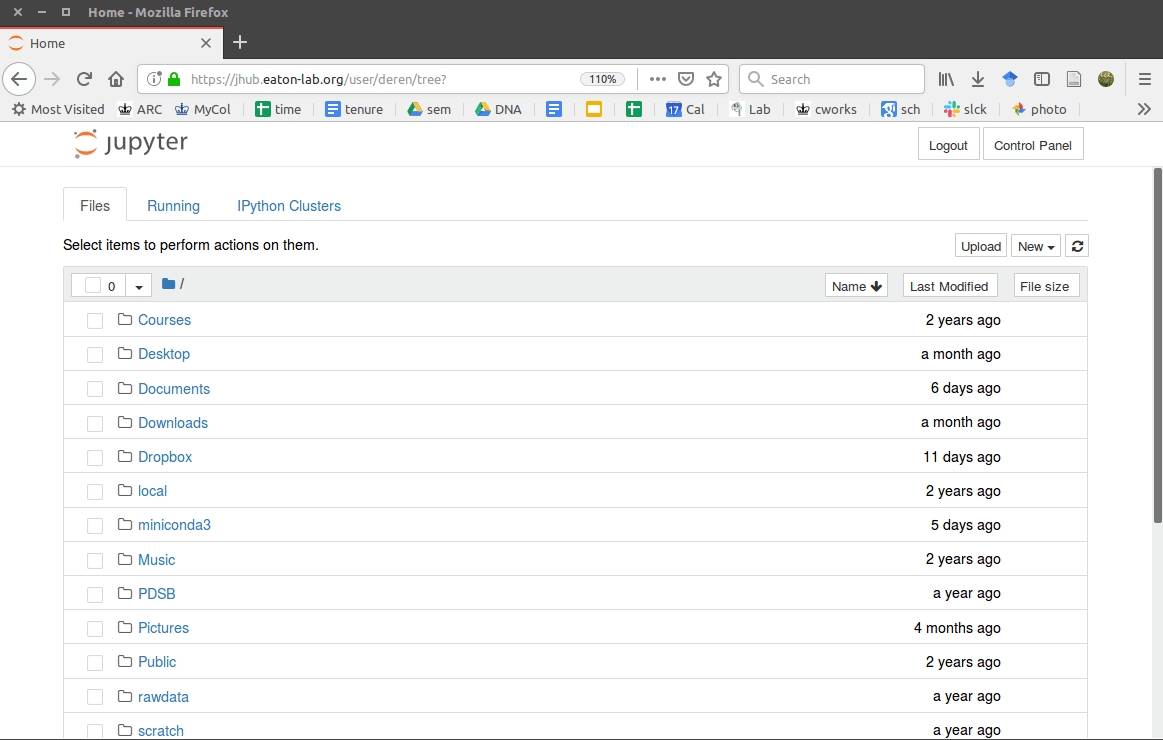
Step 8. Configure a Spawner
Here I diverge from a simpler setup in order to provide two different
spawning options, one for lab users that have a user account on the
workstation, and another for temporary users that do not have permanent
accounts. This is possible using the WrapSpawner, one of several available
spawners from jupyterhub (e.g., we already installed DockerSpawner earlier). It can be installed with pip:
In your config file you can then create multiple spawn profiles, each linked
to a different spawner setup. Here our different spawner setups include different docker images or volumes. In my setup Eaton-lab members use the dockerspawner.SystemUserSpawner which puts them in the jhub-lab3 docker image
but with access to their home directory on the system. Other temporary users
are spawned with dockerspawner.DockerSpawner as anonymous users (jovyan in
docker parlance). They will have a work directory that can persist over multiple
sessions until I eventually remove it.
Use Docker’s installation instructions to install Docker on your system. Then run the following command to make sure your docker is working.
## run the test image hello-world
docker run hello-world
We are going to want to setup two docker images: one with the basic code to
start jupyter notebooks for users, and that details all of the software that
we want to make available to users when they login. The first, called singleuser, is easy and can be downloaded with the command below. The second can also be
downloaded easily if you want to just copy my setup. I’ll detail in a later
post how I created the docker image so you can customize it.
## the image docker will use to start notebooks
docker pull jupyterhub/singleuser
## optionally also pull my docker setup
docker pull dereneaton/jhub-lab3
Finished
You can now start the jupyterhub on your workstation by running:
Users can then login by visiting your domain address:
Users will have access to a prebuilt set of software tools defined in the
docker image. They will also be located in an isolated linux system so that
they can install additional software as well, for example from
/opt/conda/bin/conda.
We have access to both the Terremoto and Habanero clusters.
Documentation for Terremoto is here, and Habanero here). On Habanero
Eaton lab members have access to 8Tb of scratch space and about 20 24-core nodes,
but these resources are shared and often busy. On Terremoto we have one
reserved 24 core node and 6Tb of scratch space, and can access all other shared
resources. On both clusters the max walltime is 5 days
(or 6 hours on the free partition, or 12 hours on the short partition).
Connecting by SSH
Use SSH from a terminal and your UNI credentials to login.
# Connect to habanero from your local computer
ssh <user>@habanero.rcs.columbia.edu
# OR, connect to terremoto from your local computer
ssh <user>@moto.rcs.columbia.edu
Setup your scratch directory
On Habanero you can access the “dsi” partition, on Terremoto use the “eaton”
partition. You can create a user specific scratch directory in the the
partition named with your UNI. This is where you should store large data files.
If you think you will need to share the data with others then use the ‘project’
space on Terremoto in the eaton directory.
# ON HABANERO# make a directory in the scratch spacemkdir /rigel/dsi/users/<user>
# make a symlink from your home dirln-s /rigel/dsi/users/<user> ~/scratch-dsi
# ON TERREMOTO# make a directory in the scratch spacemkdir /moto/eaton/users/<user>
# make a symlink from your home dirln-s /moto/eaton/users/<user> ~/scratch-user
ln-s /moto/eaton/projects ~/scratch-projects
To transfer files from your local computer to the cluster you can use scp,
or you can download data directly on the cluster if it is hosted online
somewhere. The two clusters do not share a disk space, unfortunately, so you
cannot copy data to one and access it from the other. Better to choose one
cluster for your project, probably moto.
# On your local computer# transfer files or dirs from your local computer to the scratch space
scp <path-to-file-or-dir> <user>@habanero.rcs.columbia.edu:/rigel/dsi/users/<user>
Submit jobs to the cluster using SLURM
Both clusters use the SLURM job submission system to manage shared resources on the
cluster. When you login you will be connected to the head node, which is
simply a landing pad. You should not run any intensive tasks on this node.
Instead, submit your jobs using a job script to take care of reserving
resources for your job and sending it to run on a compute node.
First we’ll make some directories to help ourselves stay organized; one
directory for job scripts and one directory for log files, which store the
output of running jobs.
# On the head nodemkdir ~/slurm-scripts/
mkdir ~/slurm-logs/
Example job submission
The header at the top of the file tells the scheduler the resources we need, which account to use (“dsi”) and how the job and output files should be named. The scripts below the header will be executed on compute node(s) once they are available. In the command below we reserve one core and simply execute the echo command to print text to the output. I name the file moto-helloworld.sh and put it in the slurm-scripts/ dir.
# open file with nano text editor on the head node
nano ~/slurm-scripts/moto-helloworld.sh
# On the head node
sbatch ~/slurm-scripts/moto-helloworld.sh
Check whether it has started yet:
# On the head node
squeue -u <user>
Once it starts check your log files for the output:
# On the head nodecat ~/slurm-logs/<jobid>.log
Start a notebook server
I do most of my work on jupyter notebooks which also provide a really nice way
to connect and work interactively on compute nodes. To start a notebook server
let’s start by generating a config file and a password. This is optional –
if you don’t set a password then a temporary token will be generated when you
start a notebook – but setting a password makes connecting a bit simpler. You
will of course need to have jupyter installed already.
# On the head node
jupyter-notebook --generate-config
jupyter-notebook password
Next let’s write a job submission script to start a notebook server. In the example below we reserve one entire node (all 24 cores by asking –exclusive). We also designate a specific port and IP to run the notebook server from. The port can be any number between 8000-9999, it is easiest if you just pick your favorite number and use it all the time. I typically use 8888 for notebooks I run locally and 9999 for notebooks I connect to remotely. The IP/hostname of the compute node is generated by the command hostname in the script.
# On the head node
nano ~/slurm-scripts/moto-jupyter-1n-1d.sh
#!/bin/sh#SBATCH --account=eaton#SBATCH --nodes=1 #SBATCH --exclusive #SBATCH --time=1-00:00:00#SBATCH --workdir=/moto/home/de2356/slurm-scripts/#SBATCH --job-name=jupyter## unset XDG variable (required when running jupyter on HPC)cd$HOMEXDG_RUNTIME_DIR=""
jupyter-notebook --no-browser--ip=$(hostname)--port=9999
Submit the job:
# On the head node
sbatch slurm-scripts/moto-jupyter-1n-1d
Check if the job has started, and take note of the hostname of the node it has connected you to.
# On the head node
squeue -u <user>
Once it starts you can connect your local computer to the notebook server running on the compute node by creating an SSH tunnel. Run the command below from your local machine, substituting in the hostname of the node that you connected to in place of the name t103. Once executed, leave this terminal window open and minimize it into the corner. You can just leave it for as long as you want to maintain the tunnel connection.
## On your local computer
ssh -N-L 9999:t103:9999 de2356@moto.rcs.columbia.edu
Now open a browser on your local computer (e.g., laptop) and enter the address localhost:9999
Interactive mode
If you only plan to do a very small amount of work it is better to just jump into
an interactive session rather than submit a job to start a notebook server or to
request many resources. This type of job will usually start quickly.
# ask for 30 min interactive session
srun --pty-t 30:00 --account=dsi /bin/bash
]]>Deren Eatonde2356@columbia.eduMaia presents at undergraduate thesis poster session2018-12-07T00:00:00+00:002018-12-07T00:00:00+00:00https://eaton-lab.org/posts/maia-poster
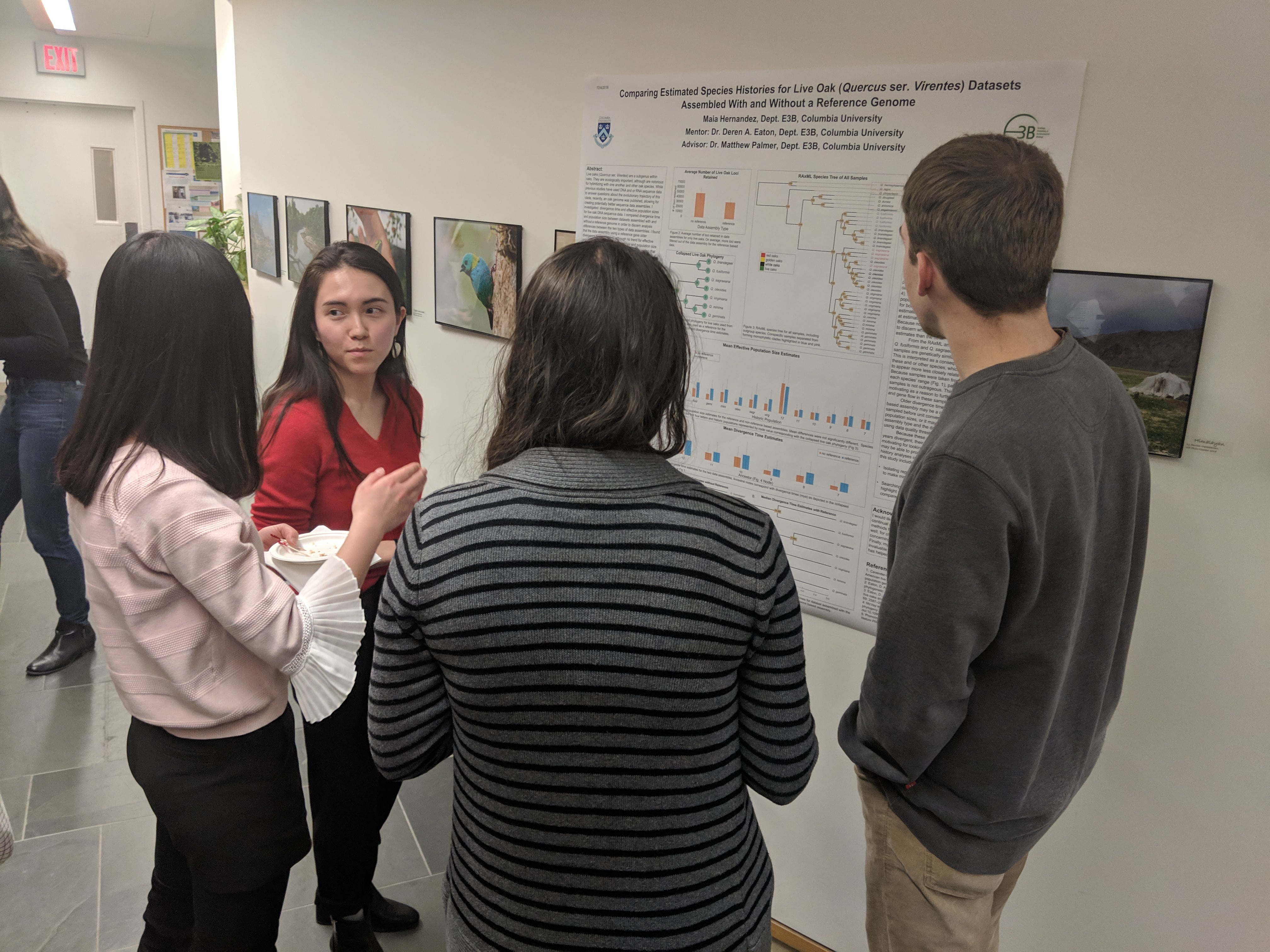
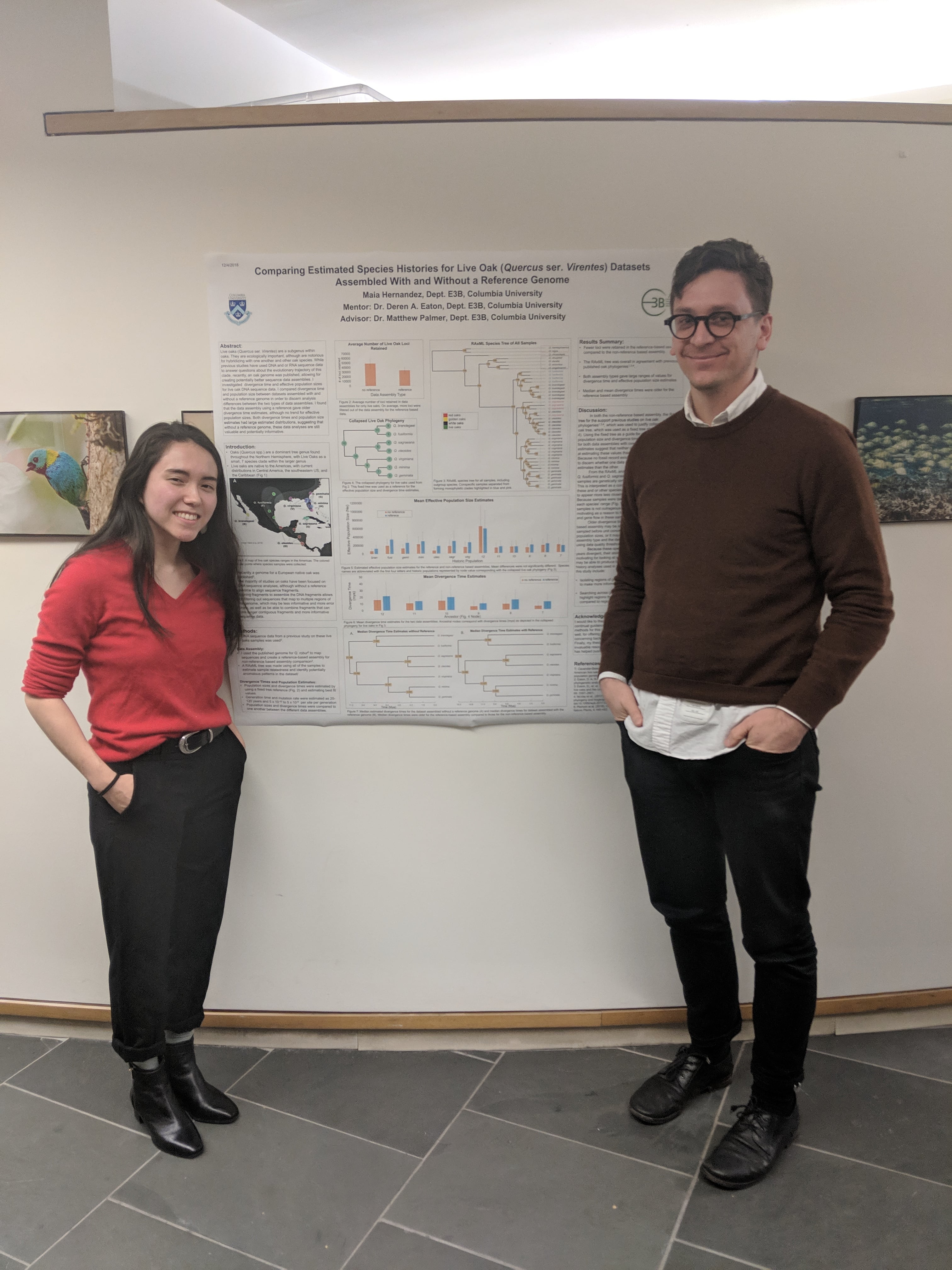
Congratulations to Maia Hernandez on presenting her thesis research at the undergraduate research poster session in E3B. Maia is studying hybridization and phylogenomics in the American live oaks using genomic RAD-seq data, and investigating bioinformatic approaches to combining these data with information from a closely related reference genome.
]]>Deren Eatonde2356@columbia.eduWelcome Jared Meek and Guo Cen to the lab!2018-09-17T00:00:00+00:002018-09-17T00:00:00+00:00https://eaton-lab.org/posts/welcome-new-lab-2
The Eaton lab is happy to welcome two new members: Jared Meek and
Guo Cen. Jared is a new M.A. student interested in plant systematics and conservation. Before even starting in the E3B program he already joined
our field expedition this summer to the Hengduan Mountains, and so he is
hitting the ground running with tons of data in hand to study phylogeography
in Pedicularis. Guo Cen is a visiting Ph.D. student from the Chinese Academy of Sciences graduate program at the Kunming Institute of Botany, and was awarded
a fellowship to study internationally. She is investigating the phylogeny and diversification of temperate
bamboo species.